
瑞士作为非欧盟成员国,其医疗器械监管体系既独立于欧盟又与国际标准高度接轨,在全球医疗领域占据着重要地位。Swissmedic是负责医疗器械注册与监管的核心机构,其审批标准严格、流程透明,尤其在创新器械和高风险医疗技术领域享有很高的国际声誉。尽管瑞士未加入欧盟医疗器械法规(MDR/IVDR),但在实际操作中广泛参考欧盟标准,并要求非瑞士制造商必须指定本地授权代表(CH-REP)协助完成注册。
监管局官网链接:https://www.swissmedic.ch/swissmedic/en/home.html
数据库链接:https://swissdamed.ch/
Medical Devices Ordinance (MedDO)
Ordinance on In Vitro Diagnostic Medical Devices(IvDO)
医疗器械(含体外诊断器械)的分类,分别遵循《医疗器械法》(MedDO)第15条和《体外诊断器械法》(IvDO)第14条。其指出分别适用于欧盟MDR和欧盟IVDR里附件VIII,与欧盟的分类规则保持基本一致。

英语,说明书标签需为瑞士官方语言(德语、法语和意大利语)其中一种。
在瑞士,医疗器械的制造商、进口商或授权代表可作为申请者提交注册申请。如果医疗器械制造商的营业地点不在瑞士,则只有在指定位于瑞士的授权代表后,其产品才可以投放市场(MedDO第51条第1款)。
根据瑞士MedDO法规及Swissdamed数据库的阶段性要求,医疗器械注册流程按产品风险分级有简化和完整两种程序,整体可分为五大核心步骤,具体如下:
1. 前期准备与主体备案:非瑞士制造商先确定产品风险等级,再必须指定瑞士授权代表(CH - REP)。
2. 技术文件筹备:I类低风险器械仅需准备基础符合性声明和产品合规文件;IIa类及以上器械要准备全套技术文档,含设计文件、ISO 13485质量管理体系证明,III类器械还得补充含瑞士人群适用性的临床评估报告。除英文文件外同时标签和说明书需提供德、法、意、任一语言版本,并标注CH - REP信息。
3. 选择路径并提交申请:有CE证书的可走简化程序,将CE证书作为核心材料提交;无CE证书则走单独国家程序,需额外提交风险 - 收益分析报告等。通过Swissmedic电子门户提交申请,还需按风险类别缴纳费用(一般为6000-20000瑞士法郎)。
4. 审核与证书获取:I类器械无需Swissmedic前置审查,2 - 4周即可完成合规备案;IIa类器械评审约90天,III类可达120天。审核中若有问题,企业需在规定时间内补正材料,审核通过后由Swissmedic颁发注册证书。
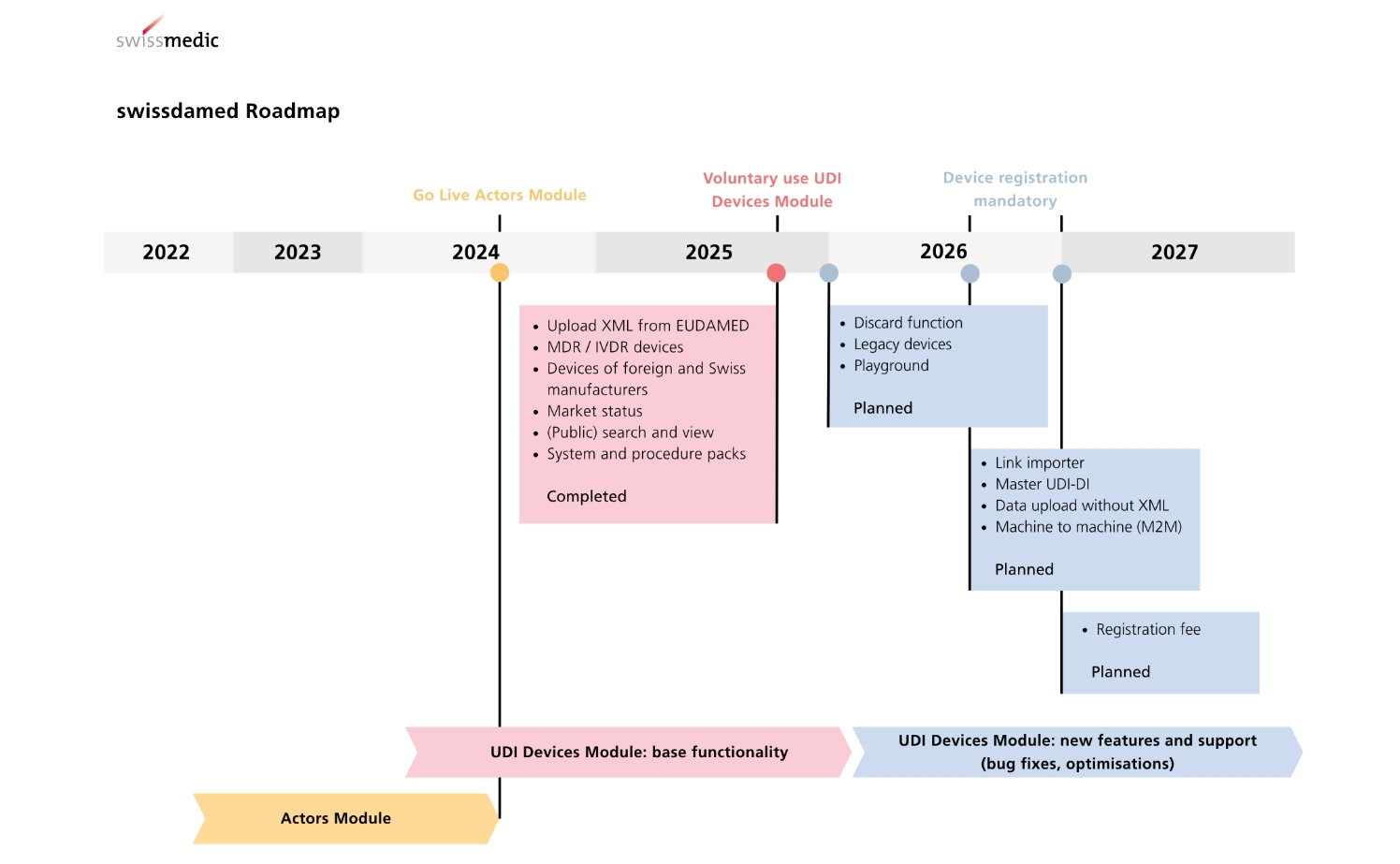
5. 器械登记与上市后监管:当前可自愿在Swissdamed的UDI模块登记器械信息,2026年7月1日起这一步将强制实施。上市后,III类器械需每年提交安全更新报告,且所有器械发生重大不良事件需24小时内通报,重大产品变更还需重新提交评审。







































