阿联酋卫生和预防部(Ministry of Health and Prevention,MOHAP)负责制定医疗器械监管的政策、法规和标准,同时也承担医疗器械的市场准入审批、质量监督等众多职责。

此外,迪拜卫生局(Dubai Health Authority,DHA)和阿布扎比卫生局(Abu Dhabi Health Authority,HAAD)等地方卫生局也在各自辖区内发挥重要的监管作用,这些机构之间相互协作确保法规的有效实施。
Medical Device Registration Guideline 2019
2019 年第 8 号法律是医疗器械监管的重要法律依据,适用于药品、医疗器械以及与健康相关的消费品
欧洲医疗器械指令(Medical Device Regulation,MDR)
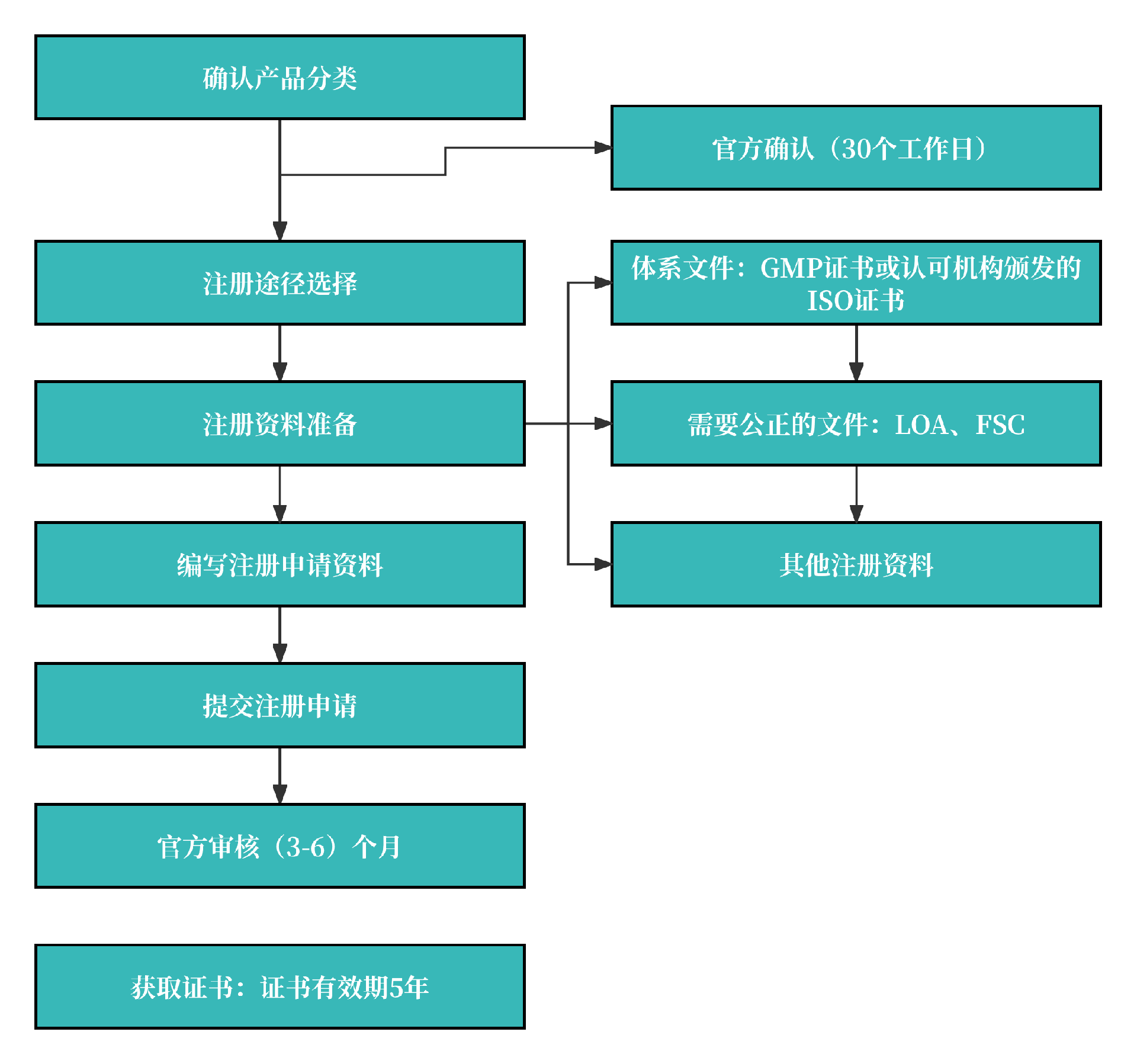
阿联酋MOHAP(阿联酋国家卫生事务部)对医疗器械的分类遵循欧洲医疗器械指令(Medical Device Regulation,MDR)和欧洲医疗器械调和标准(Medical Device Harmon),按照风险等级从低到高医疗器械分为:Ⅰ、Ⅱ、Ⅲ、Ⅳ类四个类别。体外诊断器械(IVD)分为 A、B、C、D 类。

①产品注册申请表需由公司填写、签字并盖章
②工厂有效注册证复印件
③由阿联酋大使馆认证的原产国主管部门签发的自由销售/注册证书
④公司与经销商/代表签署的授权副本
⑤质量合格证书/营销授权证书,例如 EC、510 (K)、PMA,根据设备的分类,即 I、II、III、IV 类
⑥上市后监测要求。
⑦其他国家的产品注册证复印件。
⑧产品信息,包括描述、配方、类型、尺寸、型号、配件、用途、副作用、矛盾、警告、注意事项、使用指南、包装封面照片、手册和用户手册。
⑨提供实验室要求和分析,以及某些医疗设备的定价。
⑩提供三个样品(根据设备类型)、分析证书(根据设备类型)、内外封面和手册。
⑪公司确认设备符合《医疗设备手册》(CE 符合性声明)的规格。
⑫安全性和有效性数据(针对 III、IV 类产品)。
⑬特殊要求:动物产品制造设备的合格证书。
阿拉伯语